This post continues a series (see Part 1 and Part 2) examining some features and insights when using ChemAxon Extended SMILES/SMARTS (CXSMILES) to represent repeat groups.
Repeat Groups
Inherited from CTfiles (Molfile) there are two ways to specify repeat groups in CXSMILES, you can either use use an Structure Repeat Unit (SRU) Sgroup or a Link Node. Historically link nodes are used for queries and the SRU for polymers however the boundaries are a little blurry. From a user’s perspective you probably want to handle them interchangeably – particularly if this is a query structure input. Recent versions of ChemAxon’s MarvinSketch will encode a repeat as a link node if possible. In third party tools similar interconversion is useful with a preferred (canonical) representation generated on output.
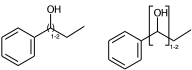
Link nodes are more limited than SRU groups but allow for a terser encoding. Firstly any crossing bonds (bonds that cross the brackets) must connect to the same repeated atom. You must also define a lower and upper bound for number of types to repeat (e.g. 1 to 3). In CTfiles the lower bound must be 1 – in practice any lower bound (including 0 is reasonable). A simple example is shown below, atom idx 1 repeats 1 to 3 times.
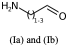
NCC=O |LN:1:1.3| (Ia)As an SRU Sgroup we can write it like this:
NCC=O |Sg:n:1:1-3:| (Ib)I have specified the subscript as “1-3” which isn’t semantically encoded but sufficient. SRUs Sgroups can have whatever you like as the subscript and you will frequently see n or m for any number of repeats but it can be anything.
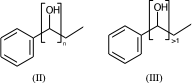
c1ccccc1C(O)CC |Sg:n:6,7:n:ht| (II)
c1ccccc1C(O)CC |Sg:n:6,7:>1:ht| (III)
The connectivity superscript of the bracket can be [head-to-tail (ht), head-to-head (hh), either-unspecified (eu)]. If the repeated part is symmetric (like in II) then it is redundant and can be omitted.
Link nodes allow you to specify the outer atoms of crossing bonds, Sgroups only support this for ladder-type polymers (as per documentation). You need the outer atoms for link nodes if there are more than two bonds – this means multiple atoms repeat. With the SRU Sgroups you just specify all the atoms that repeat. I’ve strained the brackets to demonstrate below:
c1ccccc1C(O)CC |LN:6:1.2.6.8| (IVa)
c1ccccc1C(O)CC |Sg:n:6,7:1-2:ht| (IVb)
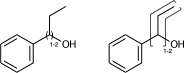
c1ccccc1C(O)CC |LN:6:1.2.6.7| (Va)
c1ccccc1C(O)CC |Sg:n:6,8,9:1-2:ht| (Vb)If the link node is in a ring, then the crossing bonds are implicitly the ones in the ring. However for portability the outer atoms should be specified whenever there is more than two bonds connected.
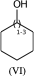
C1(O)CCCCC1 |LN:0:1.3| (VIa) acceptable
C1(O)CCCCC1 |LN:0:1.3.1.6| (VIb) preferred
C1(O)CCCCC1 |Sg:n:0,1:1-3:ht| (VIc)Something that may not be obvious when you first encounter repeat groups is that although in polymer chemistry they are typically linear, for structure queries we can also have what I will call radial repeats. With a radial repeat the repeat unit “rotates” around a single external atom – they are implicitly bounded by the number of bonds that atom can have. They can have an odd or even number of crossing bonds and it depends on the grouping of the crossing bonds as to what type of repeat you have. The table below shows this, I have put some hypothetical examples in light grey to show the pattern repeats.
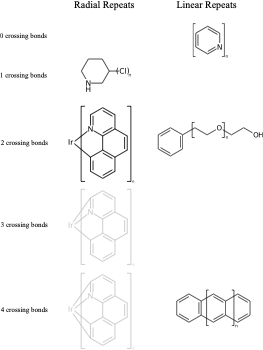
As with linear repeats, radial repeats can be represented as link nodes if the range is known and they are a single atom:
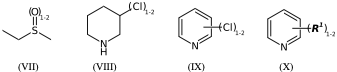
CS(=O)CC |Sg:n:2:1-2:| (VIIa)
CS(=O)CC |LN:2:1.2| (VIIb)
N1CCCC(Cl)C1 |Sg:n:5:1-2:| (VIIIa)
N1CCCC(Cl)C1 |LN:5:1.2| (VIIIb)
*Cl.n1ccccc1 |Sg:n:1:1-2:,m:0:2.3.4.5| (IXa)
*Cl.n1ccccc1 |m:0:2.3.4.5,LN:1:1.2| (IXb)
**.n1ccccc1 |$;_R1$,m:0:2.3.4.5,Sg:n:1:1-2:| (Xa)
**.n1ccccc1 |$;_R1$,m:0:2.3.4.5,LN:1:1.2| (Xb)For non ladder-type polymers CXSMILES Sgroups only capture the atoms that repeat and not the grouping of the crossing bonds. Therefore we have an ambiguity with radial spiro-repeats. A reasonable rule of thumb is if the outer atom of each crossing bond is the same then it is likely a spiro-repeat rather than linear.
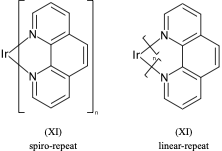
C=1C2=C3[N]([Ir][N]4=CC=CC(C=C2)=C34)=CC1 |Sg:n:0,1,2,3,5,6,7,8,9,10,11,12,13,14:n:ht| (XI)CTfiles actually have the same issue if you don’t store the coordinates. Fortunately these repeat types are relatively rare but here are some in the wild:
(US20160049599A1-20160218-C00566)
(US20150380666A1-20151231-C00746)
We extract these from patents as CXSMILES with our Text-Mining tool LeadMine (Poster).
Explicit hydrogens may be required to define the Sgroup SRU properly. If an explicit hydrogen is used care needs to be taken when suppressing explicit hydrogens. If you suppress/remove the hydrogen in (XII) the meaning changes from a PEG linear repeat to a radial repeat
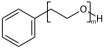
c1ccccc1CCO[H] |Sg:n:6,7,8:m:ht| (XII)Atom indices can be written in any order, ideally when writing CXSMILES you should sort the list of indices.
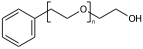
c1ccccc1CCOCCO |Sg:n:6,7,8:n:ht| (XIIa)
c1ccccc1CCOCCO |Sg:n:6,8,7:n:ht| (XIIb)
c1ccccc1CCOCCO |Sg:n:8,7,6:n:ht| (XIIc)As touched upon previously, If the repeated unit is symmetric then the subscript is irrelevant, for registration these should all be considered the same:

c1ccccc1OCOCCO |Sg:n:6,7,8:n:hh| (XIIIa)
c1ccccc1OCOCCO |Sg:n:6,7,8:n:ht| (XIIIc)
c1ccccc1OCOCCO |Sg:n:6,7,8:n:eu| (XIIIb)
c1ccccc1OCOCCO |Sg:n:6,7,8:n:| (XIIId)My interests are mainly on repeat variation for structure queries. There is a lot more that can be said on polymer registration, for example you need to canonicalise the repeat unit (frame shift). Four of these are the same structure and one isn’t:
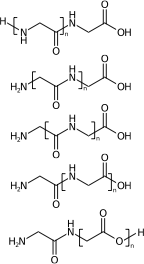
[H]NCC(=O)NCC(=O)O[H] |Sg:n:1,2,3,4:n:ht| (XIVa)
[H]NCC(=O)NCC(=O)O[H] |Sg:n:2,3,4,5:n:ht| (XIVb)
[H]NCC(=O)NCC(=O)O[H] |Sg:n:3,4,5,6:n:ht| (XIVc)
[H]NCC(=O)NCC(=O)O[H] |Sg:n:5,6,7,8:n:ht| (XIVd)
[H]NCC(=O)NCC(=O)O[H] |Sg:n:6,7,8,9:n:ht| (XIVe)A final comment is something I noticed when preparing this post. It is common in polymer chemistry to draw external external attachments as a plain bond. Here is the wikipedia depiction of polyvinyl chloride:
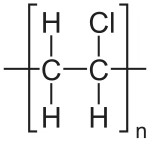
*CC(Cl)* |Sg:n:1,2,3:n:ht| (XVa)EPAM’s Ketcher looks like it automatically converts any methyl capping groups to ‘*’ on load… but this might just be a display setting. It’s possibly reasonable and it doesn’t seem to be default behaviour in the Indigo API but is an interesting auto-conversion to be aware of.
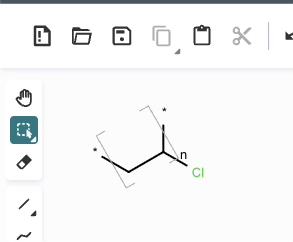
CCC(Cl)C |Sg:n:1,2,3:n:ht| (XVb)