We’ve been looking into supporting Self-Contained Sequence Representation (SCSR) in Sugar&Splice (NextMove Software’s biologics perception, conversion, and depiction toolkit, as used by PubChem). SCSR is reported (Chen et al. 2011) as a “compressed format that retains chemistry detail”.
At NextMove, we’ve long argued that the best way to store peptides for registration is as the full connection table rather than as a compressed form. The primary advantage of this is that existing infrastructure for compound registration can be reused with minimal or no changes. On modern hardware, traditional cheminformatics algorithms can easily handle much larger structures (Sayle et al. 2015). An obvious problem is that without peptide perception (e.g. using Sugar&Splice), duplicates are missed if a user inputs a fully expanded structure instead of a compressed representation. A more subtle problem emerges with modified amino-acids in compressed representations, e.g. pyroglutamic acid may be considered different it was entered as modified glutamic acid or proline.
Having distinct registration systems for peptides and compounds is more complex and therefore more error prone, and more expensive to maintain.
Formats
When I generated the SCSR output I noticed that each line for a monomer looked longer than the SMILES for each fully expanded monomer. This means that while in theory this is a compressed format, it’s actually still larger than an uncompressed SMILES string. To demonstrate here are different representations of Beefy Meaty Peptide:
FASTA:KGDEESLA
HELM:PEPTIDE1{K.G.D.E.E.S.L.A}$$$$
IUPAC Condensed:H-Lys-Gly-Asp-Glu-Glu-Ser-Leu-Ala-OH
SMILES:C[C@@H](C(=O)O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)CNC(=O)[C@H](CCCCN)N
SCSR: NextMove08101613572D 0 0 0 0 0 999 V3000 M V30 BEGIN CTAB M V30 COUNTS 8 7 0 0 0 M V30 BEGIN ATOM M V30 1 Lys 1.0 1.0 0 0 CLASS=AA ATTCHORD=(2 2 Br) SEQID=1 M V30 2 Gly 2.0 1.0 0 0 CLASS=AA ATTCHORD=(4 1 Al 3 Br) SEQID=2 M V30 3 Asp 3.0 1.0 0 0 CLASS=AA ATTCHORD=(4 2 Al 4 Br) SEQID=3 M V30 4 Glu 4.0 1.0 0 0 CLASS=AA ATTCHORD=(4 3 Al 5 Br) SEQID=4 M V30 5 Glu 5.0 1.0 0 0 CLASS=AA ATTCHORD=(4 4 Al 6 Br) SEQID=5 M V30 6 Ser 6.0 1.0 0 0 CLASS=AA ATTCHORD=(4 5 Al 7 Br) SEQID=6 M V30 7 Leu 7.0 1.0 0 0 CLASS=AA ATTCHORD=(4 6 Al 8 Br) SEQID=7 M V30 8 Ala 8.0 1.0 0 0 CLASS=AA ATTCHORD=(2 7 Al) SEQID=8 M V30 END ATOM M V30 BEGIN BOND M V30 1 1 1 2 M V30 2 1 2 3 M V30 3 1 3 4 M V30 4 1 4 5 M V30 5 1 5 6 M V30 6 1 6 7 M V30 7 1 7 8 M V30 END BOND M V30 END CTAB M END
Scaling
To test how the size of these representations scales with the peptide length, random linear unmodified peptides were generated of increasing size. The formats listed above were tested as well as the fully expanded molfile and BIOVIA generated SCSR (BIOVIA Direct 2017). The difference between the BIOVIA SCSR and the NextMove SCSR (shown above) is that the expanded template for each occurring standard amino acid is included (i.e. a monomer definition). This has a little storage overhead that varies depending on the number of unique monomers.
The results are shown below. The molfile gets reasonably large (max 500KB+), though even this could still be stored on modern hardware. The SMILES (max 16KB+) peaks just above the more condensed formats of FASTA (max 1KB), HELM (max 2KB+), and Condensed (max 4KB+).
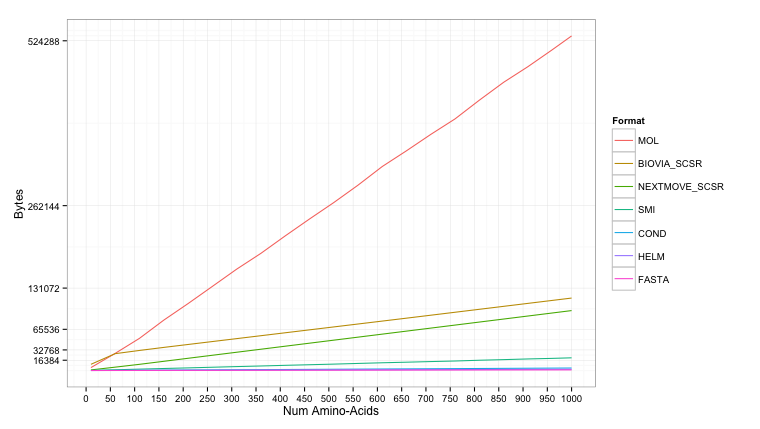
Using a log2 scale it’s easier to read the storage size:

An observation from the chart is that for small peptides the SCSR produced by BIOVIA (with monomer definitions) is actually larger than the molfile (also produced by BIOVIA). Crambin (e.g 1CRN) is often considered the boundary between a small-molecule and a protein. At 46 amino acids, it turns out that crambin reduced is smaller when stored as a fully expanded molfile compared to the SCSR representation:
| Format | Bytes |
|---|---|
| SMILES | 851 |
| SCSR (BIOVIA) | 20,448 |
| Molfile | 18,130 |
Bibliography
- Roger Sayle, John May, Noel O’Boyle. CINF 1: Generating Canonical Identifiers For (Glycoproteins And Other Chemically Modified) Biopolymers. Presented at ACS Boston Fall 2015. http://www.slideshare.net/NextMoveSoftware/cinf-1-generating-canonical-identifiers-for-glycoproteins-and-other-chemically-modified-biopolymers
- William L. Chen, Burton A. Leland, Joseph L. Durant, David L. Grier, Bradley D. Christie, James G. Nourse, and Keith T. Taylor. Self-Contained Sequence Representation: Bridging the Gap between Bioinformatics and Cheminformatics. JCIM. 2011, 51, 2186.