We’ve been busy preparing for the Boston ACS. As well as a booth (#643), we’ll be giving various talks, a poster and organising a session. Note that the CINF talk times are not necessarily correct in the PDF or printed schedule. Correct talk times and abstracts are as follows (also available online here):
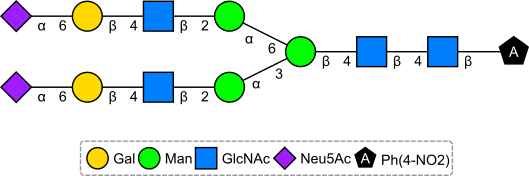
CINF 1: Generating canonical identifiers for glycoproteins and other chemically modified biopolymers
Roger Sayle, 8:35am – 9:05am, Sun, Aug 16, Room 104A – Boston Convention & Exhibition Center
Bioinformatics dogma asserts that all-atom representations, capable of encoding details such as disulfide bridging and post-translationally modified amino acids, are too unwieldy to be of practical use. In this presentation, we show how recent advances in computer power, software algorithms and storage technology require us to question this precept. We show how InChI, InChI keys and canonical SMILES can be generated for the largest known proteins, and even for nucleic acid sequences as large as viral and prokaryotic genomes. Indeed, unique identifiers derived from all-atom nucleic acid representations, allow the capture of epigenetic methylation information and circular DNA; feats that are impossible with the one-letter codes used by bioinformaticians. These unique identifiers allow the linking of mature antibodies to the unique identifiers of the plasmids used to express them. Finally, we discuss the possibility of polymer-specific implementations/optimizations of standard InChI, by showing how InChIs and InChI keys may be generated efficiently for specific classes of polymer with over a million atoms.
CINF 4: Naming algorithms for derivatives of peptide-like natural products
Roger Sayle, 10:35am – 11:00am, Sun, Aug 16, Room 104A – Boston Convention & Exhibition Center
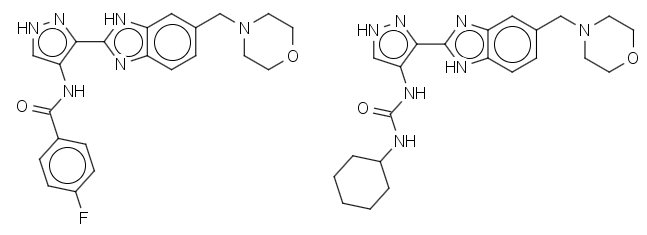
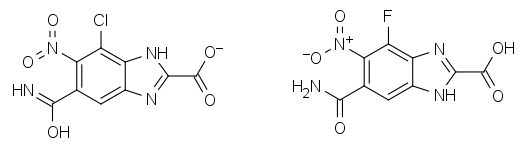
The nomenclature of natural products is a highly specialized field of biochemistry. Fortunately, some classes of natural products are more amenable to computer analysis than others. Non-ribosomal peptides and heavily post-translationally modified peptides, such as derivatives of the homodetic cycles gramicidin S and the cyclic depsi-peptide valinomycin and the natural product cyclic isopeptides anantin and sungsanpin push the current state-of-the-art in automated natural product naming. Where a compound is structurally related to an existing peptide, perceiving this relationship is required for generating succinct human understandable names. In this talk, we describe the use of databases/dictionaries based upon HELM notation and IUPAC’s condensed line notations for specifying ‘parent’ peptides from which derivatives and analogues can be named. Using the described techniques the name ‘[5-L-valine]dichotomin C’ may be assigned to the cyclic peptide CHEMBL478596. These techniques have been successfully used to identify and correct naming issues in the UniProt and IUPhar/BPS guide to pharmacology databases, which have then been updated by their curators.
CINF 18: Wikipedia and Wiktionary as resources for chemical text mining
Roger Sayle, 2:25pm – 2:45pm, Sun, Aug 16, Room 104A – Boston Convention & Exhibition Center
The resources provided by the Wikimedia Foundation provide an unprecedented resource for chemists, information professionals and natural language processing researchers in the annotation of pharmaceutically-relevant information in documents. A widely publicized example of the use of Wikipedia in artificial intelligence research is IBM’s Watson’s participation in the Jeopardy! quiz show. In this presentation, we present several chemical research applications of Wikipedia-derived data sets, including named-entity dictionaries and synonym lists for linking ontologies. The global community of volunteer contributors to these projects deserves continual recognition for the invaluable resource they enable.
CINF 29: Visualization and manipulation of Matched Molecular Series for decision support
Noel O’Boyle, 3:00pm – 3:25pm, Sun, Aug 16, Room 104B – Boston Convention & Exhibition Center
A Matched Molecular Series (MMS) is a set of molecules that differ by substitution at the same scaffold location [1]. For two molecules, this is equivalent to a Matched Molecular Pair.
We present a graphical interface for querying a database of bioactivity or physicochemical property data using a matched series. Using the database, predictions are made using the Matsy method [2] which suggests what R groups will improve the particular property value of interest.
An interesting aspect of our approach is that the interface treats the distinct R groups attached to a particular scaffold as first-class entities that can be manipulated and rearranged to see the effect on the predictions. This makes it easy, for example, to compare the predictions based simply on matched-pair information versus information from longer length series.
References:
[1] Wawer, M.; Bajorath, J. J. Med. Chem. 2011, 54, 2944.
[2] O’Boyle, N.M.; Boström, J.; Sayle, R.A.; Gill, A. J. Med. Chem. 2014, 57, 2704.
CINF 51: Analyzing success rates of supposedly ‘easy’ reactions
Roger Sayle, 10:35am – 11:00am, Mon, Aug 17, Room 104A – Boston Convention & Exhibition Center
Chemists, like insects, come in a bewildering number of varieties and specializations. Traditional retrosynthesis tools are aimed at expert synthetic chemists to assist them with challenging total syntheses, or at process chemists searching for optimal routes via obscure reaction mechanisms. In this talk, we instead consider the role of computer software to support non-experts in synthetic chemistry, such as medicinal and computational chemists. Here the challenge is not in choosing the reaction, but instead preventing silly mistakes with the most widely applied classes of named reactions. Anecdotal experience with the content of pharmaceutical ELNs shows that low yield reactions often correlate with the presence of known incompatible functional groups, such as a second halide in Suzuki couplings.
CINF 74: Unlocking chemical information from tables and legacy articles
Daniel Lowe, 2:20pm – 2:45pm, Mon, Aug 17, Room 104B – Boston Convention & Exhibition Center
Many tools for text-mining are designed to work with unstructured text. Here we present the results of our efforts to apply text-mining to the semi-structured content of tables.
We will cover the difficulties of coping with the various different ways that the contents of a table may be specified and the challenges of resolving references to elsewhere in the document. We report on the extraction of melting points, boiling points, NMR spectra and biological activity data from tabular data.
In collaboration with the Royal Society of Chemistry (RSC) we have also investigated the application of these tools to the RSC’s back archive, both in tables and free text. We cover the difficulties in adapting tools optimized for patents to journal articles and the difficulties in handling the older, less structured, text that dates from as far back as 1841.
The information extracted from this project, both from patents and the RSC’s back archive, will form a key contribution to the RSC’s public data repository. In all cases the evidence text for the extracted information is provided along with a link back to the document from which it was extracted, ensuring that the provenance of the information can be verified.
CINF 91: Chemistry enabling Chinese, Japanese, and Korean patents (Poster)
Daniel Lowe, 8:00pm – 10:00pm, Mon, Aug 17, Hall C – Boston Convention & Exhibition Center
Chinese, Japanese and Korean (CJK) patents account for over half of all national patent filings and hence are of increasing importance to patent informatics. In chemistry, searching for relevant patents relies heavily on the ability to index by chemical structures mentioned. Chemical names are typically given in the native language of the patent significantly complicating their identification and interpretation by conventional chemical text mining tools. Here we present on our approach to the translation of chemical names from CJK text and give examples of the wealth of chemical knowledge that can be unlocked.
As novel compounds are described using systematic chemical nomenclature, our approach has been developed to be especially adept at translating systematic names. Systematic chemical nomenclature in CJK languages generally follow the rules described by the IUPAC1 meaning that after translation there will exist a corresponding English name which can then be used with conventional chemical text mining tools.
Strategies for translation vary between languages. In Chinese each morpheme of an English chemical name is represented by one or more Hanzi. The interpretation of a Hanzi may be context dependent which is handled by looking at the environment in which it occurs. Japanese and Korean chemical names, by contrast, are mostly transliterations of English/German chemical nomenclature into Katakana and Hangul respectively.
As a case study we applied our approach to 44 thousand Korean patents (1990-2013) that were likely to contain chemistry and extracted 1.5 million distinct compounds. 177 thousand of these compounds were not found by a comparable analysis of US patents. Of the 759 thousand compounds, first disclosed between 2006 and 2013 by both a US and a Korean patent, for 362 thousand the Korean patent was published earlier.
CHAS 31: Encoding reactive chemical hazards and incompatibilities in an alerting system
John May, 2:30pm – 2:50pm, Tue, Aug 18, Waterfront 1A/1B – Seaport Hotel and World Trade Center
Of the many chemical reactions performed by synthetic chemists in the pharmaceutical industry and academia, some are potentially more hazardous than others. Fortunately, best practices, compliance and education helps ensure that incidents are rare, but as highlighted by the recent explosion and building evacuation at two UK universities in March 2015, constant vigilance is necessary to ensure a safe work environment. The primary problem is not that chemical safety information, for example from MSDS/SDS data sheets, Bretherick’s Handbook or the internet, is readily available, but that the volume of such information makes it difficult for an experimentalist to identify relevant risks in a timely manner.
In this talk, we describe our attempts to encode the Environmental Protection Agency’s (EPA’s) guidance entitled ‘A Method for Determining Compatibility of Hazardous Waste’, 1980, in an XML file format. Typical current state-of-the-art methods for alerting potential chemical safety hazards, for example in ELNs, simply annotate reactants with codes extracted from their MSDS/SDS data sheets, such as Global Harmonized System of Classification and Labelling of Chemicals (GHS) or EU R-phrases/S-phrases, leaving the chemist to manually assess whether any of the described incompatibilities is relevant. In this work, we use combinations of SMARTS patterns (for chemical classes) and InChIs (for specific molecules), to capture known reagent incompatibilities, that may be safe in isolation. Specific alerts describe documented incompatibilities between compounds (e.g. acetone and H2O2) while more general alerts can capture known or inferred incompatibilities between functional groups (e.g. ketones and peroxides). The encoding is hierarchical allowing only the most relevant warnings to be triggered. Alerts are encoded in a flexible XML format, facilitating extension and exchange. Advanced features of this XML format, include the ability to specify reactant quantities, and the use of predicted properties (such as of products) in rules.
CINF 141: So I have an SD File…what do I do next?
Rajarshi Guha and Noel O’Boyle, 1:35pm – 1:55pm, Wed, Aug 19, Room 104A – Boston Convention & Exhibition Center
Cheminformatics tasks cover a wide range of topics, from manipulating chemical structure file formats to predicting properties of chemical structures. The common theme underlying all these tasks is the handling of chemical structures. Yet frequently key aspects of structural information are lost, altered or ignored during even the most routine of processing tasks either through a misunderstanding of how tools work, limitations of the tools used or unfamiliarity with the features (or lack thereof) of particular chemical file formats.
Here we present a compendium of the “Dos and Don’ts of cheminformatics”. Using examples drawn from over a decade of involvement with open source cheminformatics toolkits [1] [2] and a variety of cheminformatics applications, as well as from recent commentaries on chemical structure databases, we illustrate some misconceptions regarding how chemistry data is stored, propose best practices for preserving chemical information intact, and end with a cautionary suggestion: “don’t trust, but verify”.
References:
[1] Steinbeck, C. et al., J.Chem. Inf. Comput. Sci., 2003, 43, 493-500
[2] O’Boyle, N.M. et al., J. Cheminf., 2011, 3, 33






 The Journal Impact Factor (or JIF) is the mean number of citations to articles published in a journal in the previous 2 years. Now, the mean is often a good measure of the average but not always. To decide whether it’s a good measure, it is often sufficient to look at a histogram of the data. The image above from a
The Journal Impact Factor (or JIF) is the mean number of citations to articles published in a journal in the previous 2 years. Now, the mean is often a good measure of the average but not always. To decide whether it’s a good measure, it is often sufficient to look at a histogram of the data. The image above from a 


{kind=link}
{kind=link}