Chemical structure diagrams are essential in describing and conveying chemistry. Extracting chemistry from documents using text-mining (see NextMove Software’s LeadMine) is extremely useful but will miss anything described only by an image.
As a general approach to mining chemistry from images, one may consider using image-to-structure programs such as: OSRA, CliDE, ChemOCR, and Imago OCR. However, image-to-structure is not easy or quick and can be prone to compounding errors (e.g. OCR).
At NextMove we approach this problem slightly differently. It turns out that in some cases the source sketch files used to create the chemical diagrams may be available and provide a ‘cleaner’ data source than the raster images.
Although the data is ‘cleaner’ in terms of digital representation, naïvely exporting the connection table stored in a sketch file can lead to artificial and erroneous structures. The main problems stem from the stored representation (connection table) imprecisely reflecting what is displayed. To account for these issues, the NextMove Software converter (code name: Praline) applies correction, interpretation, and categorisation to sketches. The transformed connection table (currently written as ChemAxon Extended SMILES [CXSMILES]) better reflects what is actually displayed.
Let’s take a look at what’s possible with three examples:
1) US 2015 344500 A1
Method 9 in US 2015 344500 A1 describes a four step synthesis:
Using image-to-structure SureChEMBL extracts four structures, I’ve added the titles to make it easier to pair up:
Compound 2-2 was not correctly extracted and looks like OCR has mistakenly recognised the -OBn as -OBu. The flurobenzene probably comes from Compound 9-1 where the label (Boc)2N- is difficult to recognise. The products of Step 4 contain valence errors and were probably thrown out as a recognition error.
However, by reading the ChemDraw files directly it’s possible to extract everything “warts and all”. To process this sketch the key interpretation phases are:
- Line Formula Parsing – Using a strict yet comprehensive algorithm condensed labels are corrected and expanded.
- Reaction Role Assignment – The reaction scheme layout is common to patents and made easier by looking for the USPTO-specific ‘splitter’ tag. To make valid reactions, reactants are duplicated and added to the previous step.
- Agent Parsing – Based on the location the complete label “Boc2, DIEA” can be correctly processed. Agents can be a mix of trivial names, systematic names and formulas.
- Clear Ambiguous Stereochemistry – One of the hashed wedges in Compounds 9-1, 9-2, and 9-3 is poorly placed between two stereocenters. In the stored representation both stereocentres are defined but we remove the definition at the wide end of the wedge.
- Category Assignment – Based on the content we tag the output with a category for quick filtering. This is described more in the poster (see below).
Here are the results of our extraction, categorised as specific reactions:

C1=CC=C(C(=C1)[C@]2(N=C(C(S(C2)(=O)=O)(C)C)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)COCC3=CC=CC=C3)F>[Li]CCCC.CC=O>C1=CC=C(C(=C1)[C@]2(N=C(C(S(C2C(C)O)(=O)=O)(C)C)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)COCC3=CC=CC=C3)F

C1=CC=C(C(=C1)[C@]2(N=C(C(S(C2C(C)O)(=O)=O)(C)C)N(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C)COCC3=CC=CC=C3)F>B(Br)(Br)Br.C(Cl)Cl>C1=CC=C(C(=C1)[C@]23NC(C(S([C@@H]3C(OC2)C)(=O)=O)(C)C)=N)F

C1=CC=C(C(=C1)[C@]23NC(C(S([C@@H]3C(OC2)C)(=O)=O)(C)C)=N)F>CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.CCN(C(C)C)C(C)C>C1=CC=C(C(=C1)[C@]23N=C(C(S([C@@H]3C(OC2)C)(=O)=O)(C)C)NC(=O)OC(C)(C)C)F
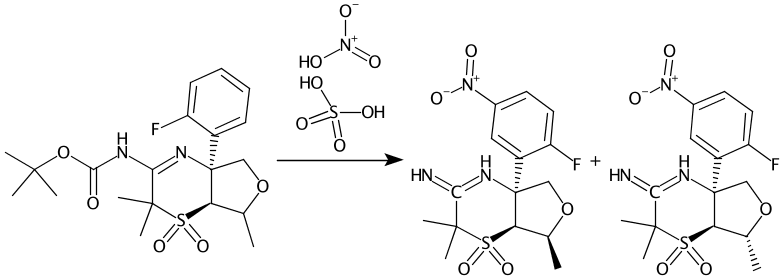
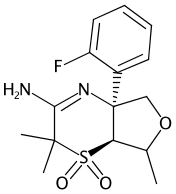
C1=CC=C(C(=C1)[C@]23N=C(C(S([C@@H]3C(OC2)C)(=O)=O)(C)C)NC(=O)OC(C)(C)C)F>[N+](=O)(O)[O-].OS(=O)(=O)O>C1(=CC=C(C(=C1)[C@]23[NH]=C(C(S([C@@H]3[C@@H](OC2)C)(=O)=O)(C)C)=N)F)[N+](=O)[O-].C1(=CC=C(C(=C1)[C@]23[NH]=C(C(S([C@@H]3[C@H](OC2)C)(=O)=O)(C)C)=N)F)[N+](=O)[O-]
Compounds 2-2 and 9-1 are now correctly extracted and actually novel to PubChem. We don’t try to correct author errors and so the bad valence is also preserved as drawn in Step 4.
2) US 7092578 B2
US 7092578 B2 is not a chemical patent but does have ChemDraw files. Here ChemDraw has been misused to draw tables, and direct export results in a cyclobutane grid. These are a well known class of bad structures in PubChem and have been referred to as chessboardanes. In addition to extracting the chemical structure, Praline assigns a categorisation code. This allows us to flag structures with potential problems as well as those with no real chemistry at all.
Resulting PubChem Compound CID 21040251:

C1C2C3C4C5C6C7C8CC9C%108C%117C%126C%135C%144C%153C%162C1C%17C%18%16C%19%15C%20%14C%21%13C%22%12C%23%11C%24%10C9C%25C%26%24C%27%23C%28%22C%29%21C%30%20C%31%19C%32%18C%17C%33C%34%32C%35%31C%36%30C%37%29C%38%28C%39%27C%40%26C%25C%41C%42%40C%43%39C%44%38C%45%37C%46%36C%47%35C%48%34C%33C%49C%50%48C%51%47C%52%46C%53%45C%54%44C%55%43C%56%42C%41C%57C%58%56C%59%55C%60%54C%61%53C%62%52C%63%51C%64%50C%49CC%64C%63C%62C%61C%60C%59C%58C%57
Praline assigns the category No Connection Table and so it can be easily ignored.
3) US 6531452 B1
Strange connection tables don’t just come from non-chemistry patents, US 6531452 B1 like many chemistry patents contain a generic (Markush) claim. Earlier we saw the label -OBn misread by OCR. Even without OCR a condensed label may be expanded wrongly in the underlying representation, particular if the structure is generic.
“…at least one of R2 and R3 is”
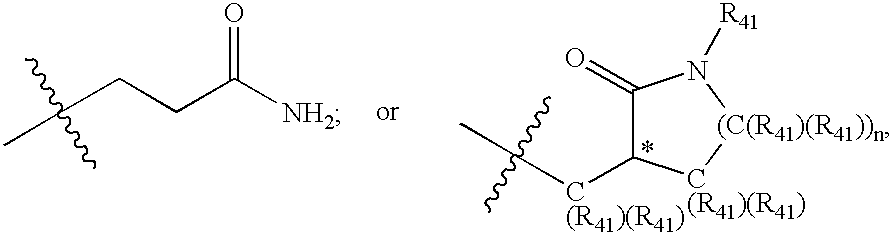
In PubChem you’ll find the compound CID 22976968 has been extracted from this sketch:

C1C2C13C24C35C46C57C68C79C81C92C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C22C11C2C1
Where did it come from? Well it turns out the generic label >C(R41)(R41) has been automatically expanded and stored in the file as:
Somewhere the Rs have been promoted to carbons and submitted to PubChem. Praline recognises and interprets generic labels and the attachment points (drawn here as tert-butyl) and categorised the sketch as a generic substituent. Here’s the output:
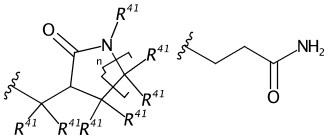
C1(C(C(C(N1*)(*)*)(*)*)C(*)(*)*)=O.*CCC(N)=O |$;;;;;R41;R41;R41;R41;R41;;_AP1;R41;R41;;_AP1$,Sg:n:3,6,7:n:ht|
Conclusion
Image-to-structure is slow; due to this, SureChEMBL currently has only processed images using image-to-structure from 2007 onwards (Papadatos G. 2015). In contrast Praline can process the entire archive of US Patent Applications and Grants with more than 24 million ChemDraw files (2001 onwards) in only 5 hours (single threaded).
Although the naïve molfile exports from the ChemDraw sketches are provided by the USPTO they have less information than the source ChemDraw sketch file. Reading the pre-exported molfile is significantly less accurate than interpreting the ChemDraw sketch, and even image-to-structure often produces more accurate results.
Other than U.S. Patents, this technology can be applied to sketch files extracted from Electronic Lab Notebooks (see NextMove Software’s HazELNut) as well as Journals where the publishers have held on to the sketch file submissions.
At the upcoming ACS in Philadelphia, Daniel will be presenting how some structures can only be extracted when the output from text-mining and sketches are combined. “The whole is greater than the sum of the parts” – Aristotle.
A poster on this work was presented at the 7th Joint Sheffield Conference on Chemoinformatics: