My earlier “On the other hand” blog post considered some of the issues of representing D- amino acids. In this post, I discuss the representation of amino acids with sidechain stereochemistry in nomenclature and peptide registration systems. Handling of chiral sidechains is potentially tricky and non-trivial, as indicated by the Pistoia Alliance’s HELM editor which restricts the user to only 17 (of the 19) standard D-form amino acids, explicitly prohibiting the specification D-threonine and D-isoleucine.
My earlier “On the other hand” blog post considered some of the issues of representing D- amino acids. In this post, I discuss the representation of amino acids with sidechain stereochemistry in nomenclature and peptide registration systems. Handling of chiral sidechains is potentially tricky and non-trivial, as indicated by the Pistoia Alliance’s HELM editor which restricts the user to only 17 (of the 19) standard D-form amino acids, explicitly prohibiting the specification D-threonine and D-isoleucine.
Threonine (Thr) and Isoleucine (Ile)
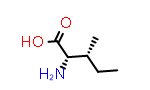
The most frequently encountered cases of sidechain stereochemistry occur in the naturally occurring amino acids threonine and isoleucine, which each contain a chiral carbon atom at their beta carbon position.
 |
L-Thr aka (2S,3R) |
PDB Code: THR |
CID6288 |
 |
L-Ile aka (2S,3S) |
PDB Code: ILE
| CID6306 |
By convention, the D-forms of these amino acids flip both stereocenters.
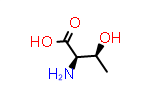 |
D-Thr aka (2R,3S) |
PDB Code: DTR |
CID69435 |
 |
D-Ile aka (2R,3R) |
PDB Code: DIL |
CID76551 |
The forms of these amino acids where just the sidechain stereochemistry is inverted are referred to as “allo-” forms, allothreonine (written aThr or alloThr) and alloisoleucine (written aIle or alloIle).
 |
L-aThr aka (2S,3S) |
PDB Code: ALO |
CID99289 |
 |
D-aThr aka (2R,3R) |
PDB Code: 2TL |
CID90624 |
 |
L-aIle aka (2S,3R) |
PDB Code: IIL |
CID99288 |
 |
D-aIle aka (2R,3S) |
PDB Code: ??? |
CID94206 |
Things really get interesting when stereochemistry is unspecified (either a racemate or unresolved chiral center) at either of these stereocenters. This is not uncommon when working with SMILES strings or MOL files, but almost always indicates some loss of information as the biology/chemistry will nearly universally refer to one of the four fully specified steroisomers above.
Perhaps the easiest case to denote is the case of unspecified tetrahedral stereochemistry at the alpha carbon position, for which the “DL-” prefix is conventionally used.
A less widely appreciated convention, is the use of the Greek letter xi (ξ) in amino acid and natural product nomenclature, for chiral centers of unknown configuration (3AA-4.5). Here I propose the use of the prefix “xi” or “xi-” in an identical way to “allo” or “allo-” to produce xi-threonine (xiThr) and xi-isoleucine (xiIle) when the beta
carbon stereochemistry is undefined/unspecified.
4-Hydroxyproline, Hyp
An example of a non-natural (but frequently occurring) amino acid with sidechain stereochemistry is “4-hydroxyproline”. Here the symbol Hyp is understood to refer to the more common trans- form, so the prefix “cis” or “cis-” is use to refer to the alternate configuration, such as the symbol “cis-Hyp”.
 |
L-Hyp aka (2S,4R) |
PDB Code: HYP |
CID5810 |
 |
L-cisHyp aka (2S,4S) |
PDB Code: HZP |
CID440015 |
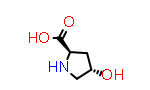 |
D-Hyp aka (2R,4S) |
PDB Code: ??? |
CID440074 |
 |
D-cisHyp aka (2R,4R) |
PDB Code: ??? |
CID440014 |
Once again unspecified configurations at the alpha- and gamma- carbon locants of Hyp can be described by “DL-” and “xi-” prefixes as before.
Note that although a few sources refer to names such as “cis-D-Hyp”, it is more usual to order terms consistently (where possible) with the “D-“, “L-” or “DL-” prefix at the start and the “allo”, “xi”, “cis”, “nor” and “homo” prefixes adjacent to the three-letter code.
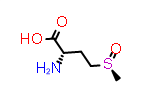
Methionine sulfoxide, Met(O)
A simpler case of sidechain stereochemistry occurs when the amino acid name doesn’t imply a default stereochemistry. In these cases, the usual Cahn, Ingold and Prelog (CIP) rules can be used to assign R and S (or E and Z) descriptors appropriately. A simple example of this is methionine sulfoxide, which is commonly represented by the symbol “Met(O)”. In this case, the sulfur atom bearing the substitution may adopt one of two configurations requiring a “R-” or “S-” prefix to the substituent suffix.
Image credit: EmsiProduction on Flickr